Nuovo studio italiano sulle malattie mitocondriali
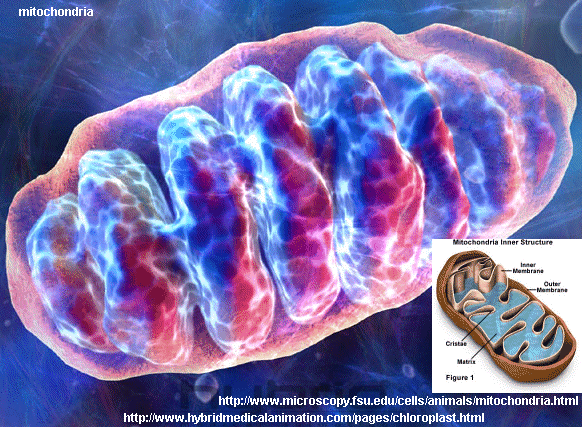
Mitocondrio
Cataratta congenita, iposviluppo somatico e miopatia mitocondriale: queste tre diverse patologie sarebbero accomunate da un`origine simile che scaturisce da alcune mutazioni del gene GFER (augmenter of liver regeneration ERV1 homolog, anche noto come Augmenter of Liver Regeneration). La scoperta tutta italiana, effettuata dai ricercatori del “Centro Dino Ferrari”- Università degli Studi di Milano e Fondazione Irccs Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena e in pubblicazione sul The American Journal of Human Genetics, permette di aggiungere un nuovo tassello allo studio delle cause e dei meccanismi delle patologie mitocondriali – caratterizzate da esordio infantile e da progressioni gravi e invalidanti – aprendo la porta a future terapie anche per malattie come la Sclerosi laterale amiotrofica.
La ricerca – La proteina codificata dal gene GFER ricopre un ruolo fondamentale nell’importo all`interno dei mitocondri di una classe di piccole proteine di primaria importanza: tra di esse, infatti, sono diverse quelle essenziali per l’organizzazione e l’assemblaggio dei componenti della catena respiratoria, nonché per i processi di detossificazione (come il trasportatore del gene SOD1, le cui mutazioni causano una forma di Sclerosi Laterale Amiotrofica Familiare). È studiando una famiglia in cui tre dei cinque figli presentano cataratta congenita, iposviluppo somatico e miopatia mitocondriale, ad esordio infantile, che Alessio Di Fonzo in collaborazione con il gruppo di ricerca diretto da Giacomo Comi ha scoperto che questa condizione familiare è causata da alcune mutazioni nel gene GFER: “È la prima volta che questo processo di importo delle proteine nello spazio intermembrana mitocondriale viene direttamente coinvolto nella patogenesi di una malattia umana – si legge nella ricerca – ed è verosimile che lo studio di GFER e delle altre proteine coinvolte in questo meccanismo potrà chiarire presto la causa genetica di numerose altre malattie mitocondriali e neurodegenerative”.
I mitocondri: piccoli, ma indispensabili – I mitocondri sono organuli presenti all’interno di ogni cellula dell’organismo umano. Più del 90% dell’energia utilizzata dal nostro corpo è prodotta in una complessa serie di reazioni biochimiche che avvengono a livello della catena respiratoria mitocondriale, che è formata da un`articolata rete di proteine che vengono assemblate sulla base di istruzioni presenti sia nel DNA mitocondriale, sia in numerosi geni del DNA nucleare.
Se non funzionano a dovere? – Un difetto nella funzionalità mitocondriale, riducendo l’attività metabolica ossidativa, porta alla compromissione dei livelli energetici di cellule, tessuti e interi organi, definendo uno stato patologico comunemente indicato con il termine “mitocondriopatia”. Sotto questo nome sono comprese condizioni eterogenee in cui si osserva il coinvolgimento di specifici tipi cellulari o, più spesso, l’interessamento multisistemico con compromissione di diverse componenti del sistema nervoso centrale, degli organi neurosensoriali, dell’apparato endocrino, del tessuto cardiaco ed epatico, nonché dell’apparato gastrointestinale ed ematopoietico.
L’ereditarietà materna
Come regola generale, tutti i mitocondri dello zigote derivano dall’ovulo (quelli contenuti nello spermatozoo vengono in quasi tutti i casi distrutti dopo la fecondazione).

Quindi una madre con mutazioni del DNA mitocondriale le trasferisce a tutta la sua progenie, ma solo le sue figlie potranno a loro volta trasferire la mutazione.
Questa regola non viene seguita sempre: uno studio recente ha infatti dimostrato in un paziente affetto da una miopatia mitocondriale la trasmissione paterna di DNA mitocondriale nel muscolo scheletrico (ma non in altri tessuti).
Le mitocondriopatie – Diverse le patologie più gravi che derivano dal malfunzionamento dei mitocondri: l’encefalopatia mitocondriale con ictus (MELAS) ed epilessia mioclonica (MERRF), la sindrome di Kearns-Sayre (KSS), la sindrome di Pearson, le sindromi neonatali ed infantili di Leigh e Alpers, l’encefalopatia con neuropatia e disturbi gastrointestinali (MNGIE), la neuropatia con atassia e disartria (SANDO) e le sindromi da deplezione del DNA mitocondriale. La frequenza di queste malattie è stata solo parzialmente stimata in alcune popolazioni: un recente studio inglese identifica mutazioni potenzialmente patogeniche nel DNA mitocondriale in un bambino su 200 nati vivi. Se a queste mutazioni si aggiungono quelle presenti in numerosi geni del DNA nucleare, appare evidente che i disordini mitocondriali nel loro insieme sono eventi tutt’altro che rari. Nonostante l`estensione delle conoscenze genetiche in merito alle mitocondriopatie acquisite negli ultimi anni, la causa delle malattie – ad esordio infantile con progressioni gravi ed invalidanti – rimane ancora sconosciuta nella maggioranza dei casi.
La ricerca “The Mitochondrial Disulfide Relay System Protein GFER Is Mutated in Autosomal Recessive Myopathy with Cataract and Combined Respiratory Chain Deficiency” verrà pubblicata sull`American Journal of Human Genetics ( http://www.cell.com/AJHG/home ) il 15 maggio 2009.
Alte fonti:
www.mitopedia.org/home.htm
Azione di un mitocondrio
I mitocondri
www.iprase.tn.it